Filter
1. Lipinski's Rule of Five (Read More)
This rule helps to predict if a biologically active molecule is likely to have the chemical and physical properties to be orally bioavailable. The Lipinski rule bases pharmacokinetic drug properties such as absorption, distribution, metabolism and excretion on specific physicochemical properties such as:
Hydrogen Bond Donors ≤ 5
Hydrogen Bond Acceptors ≤ 10
Molecular Mass ≤ 500 Da
Partition Coefficient ≤ 5
References:
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Revs. 1997;23:3–25.
2. QED (Read More)
The QED stands for quantitative estimate of druglikeness and it reflects the underlying distribution of molecular properties including molecular weight, logP, topological polar surface area, number of hydrogen bond donors and acceptors, the number of aromatic rings and rotatable bonds, and the presence of unwanted chemical functionalities.
References:
Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012 Jan 24;4(2):90-8.
3. REOS (Read More)
REOS stands for Rapid Elimination of Swill, it filters problematic functional groups that may lead to false positives due to reactivity or assay interference, which have long been noted as a problem in HTS efforts. Examples of some problematic functional groups:
References:
Huggins DJ, Venkitaraman AR, Spring DR. Rational methods for the selection of diverse screening compounds. ACS Chem Biol. 2011 Mar 18;6(3):208-17.
4. Veber (Read More)
Veber’s filter includes:
Number of Rotatable Bonds ≤ 10
Total Polar Surface Area ≤ 140
References:
Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623.
5. Ghose (Read More)
Ghose’s filter includes:
160 ≤ Molecular Weight ≤ 480
−0.4 ≤ WLOGP (Lipophilicity) ≤ 5.6
40 ≤ Molar Refractivity ≤ 130
20 ≤ Number of Atoms ≤ 70
References:
Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68.
6. Drug Like (Read More)
7. Rough Similar Properties
This filter returns molecules with molecular weight, logP, number of heavy atoms, and number of rings between -10% and +10% to the query molecule.
8. Murcko Scaffold
The Murcko scaffold contains information about the backbone of a molecule and it is obtained by extracting the ring structures and the linkers that connect them.
9. Substructure
When a structure is present in a bigger chemical structure, then the former structure is referred as a substructure of the latter. Example: Diclofenac is a substructure of Aceclofenac.
10. Superstructure
When a structure is present in a bigger chemical structure, then the latter structure is referred as a superstructure of the former. Example: Diclofenac is a superstructure of Benzene.
11. No Solvents
This filter strips off the solvent molecules from the resultant molecules.
12. No Rare Atoms
This filter eliminates structures containing:
Elements with Atomic Number > 36
13. Regioisomers (Read More)
This filter eliminates isomers that differ only on the position of a functional group, substituent, or some other feature on a "query" structure.
14. PAINS (Read More)
PAINS stands for Pan-Assay Interference Compounds, this filter eliminates compounds that produce false positives in high-throughput compound screens.
References:
Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740.
15. Murcko Scaffold Hop
This filter returns molecules with similar Murcko Scaffold and is useful for scaffold replacement.
Sort
This can sort the resultant molecules based on the selected similarity metric.
2. Morgan Tanimoto (Read More)
Morgan Tanimoto is the tanimoto similarity between Morgan fingerprints. The bits in the Morgan fingerprint corresponds to the circular environments of each atom in a molecule. The number of neighboring bonds and atoms to consider is set by the radius (the default is 3).
3. MACCS (Read More)
The MACCS (Molecular ACCess System) keys are one of the most commonly used structural keys. There are 166 SMARTS patterns corresponding to the MACCS keys. In the MACCS keys, the structure of a molecule is encoded into a binary bit string (that is, a sequence of 0’s and 1’s), each bit of which corresponds to a “pre-defined” structural feature (e.g., substructure or fragment). If the molecule has a pre-defined feature, the bit position corresponding to this feature is set to 1 (ON). Otherwise, it is set to 0 (OFF).
4. ERG (Read More)
ERG stands for Extended Reduced Graph and it is an extension of reduced graphs. ERG groups atomic substructures together based on shared characteristics (such as hydrogen-bonding, ring systems, and pharmacophoric characteristics). ERG also includes specific modifications to better represent the pharmacophoric size, characteristics, and shape of the molecules.

References:
1. Stiefl N, Watson IA, Baumann K, Zaliani A. ErG: 2D pharmacophore descriptions for scaffold hopping.
J Chem Inf Model. 2006 Jan-Feb;46(1):208-20.
2. Garcia-Hernandez C, Fernández A, Serratosa F. Ligand-Based Virtual Screening Using Graph Edit Distance as Molecular Similarity Measure.
J Chem Inf Model. 2019 Apr 22;59(4):1410-1421.
5. USR (Read More)
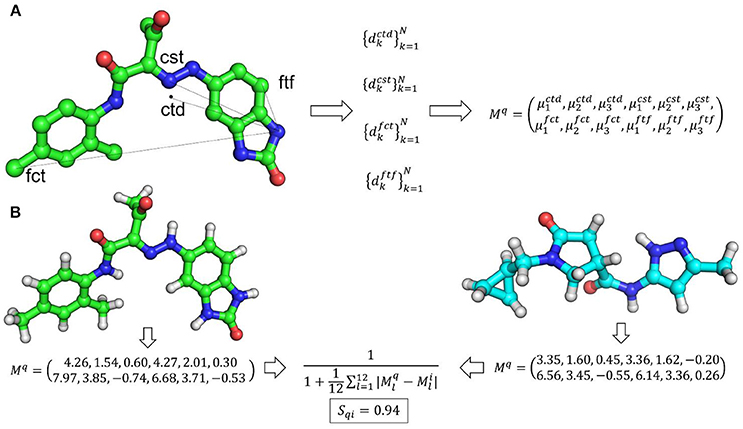
Ultrafast shape recognition (USR) is a recent shape similarity technique. USR calculates the distribution of all atom distances from four reference positions: the molecular centroid (ctd), the closest atom to molecular centroid (cst), the farthest atom from molecular centroid (fct) and the atom farthest away from fct (ftf). Consecutively, the first three statistical moments (mean, variance, and skewness of distribution) are calculated from each of these distributions. Hence, each molecule has a vector of twelve descriptors to describe its 3D shape. Finally, the similarity between shapes of two molecules is calculated through an inverse of the Manhattan distance of these 12 values. Mq and Mi are vectors of shape descriptors for query and lth molecule, respectively.
References:
Kumar A, Zhang KYJ. Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery.
Front Chem. 2018 Jul 25;6:315.