API Tutorial
In this tutorial, we will learn how to use the API to interact with CHEESE platform. We will cover the following topics: - How to retrieve molecules from available databases - Search and batch search - Comnputing CHEESE Embeddings - Computation of similarities
Prerequisities
- Python
- NumPy (optional):
pip install numpy - RdKit (optional):
pip install rdkit - Rich (optional):
pip install rich
import requests
import json
from rdkit import Chem
from rdkit.Chem import Draw
import numpy as np
from rich.pretty import pprint
np.set_printoptions(precision=3)
Sanity Check
Public API
MY_URL = "https://api.cheese.deepmedchem.com"
API_KEY = "eyJhbGciOi..." # Obtained by signing up at https://cheese.deepmedchem.com/ and clicking on "Generate API Key"
headers = {"X-API-Key": API_KEY, "accept": "application/json" }
On-Prem API
MY_URL = "http://cheese-database.ch.themama.ai:9002" # URL of the API, change it if you are using a different one
headers = {'accept': 'application/json'} # headers for the request, we want a JSON response
Test Request (Health Check)
{'message': 'Health check successful !!'}
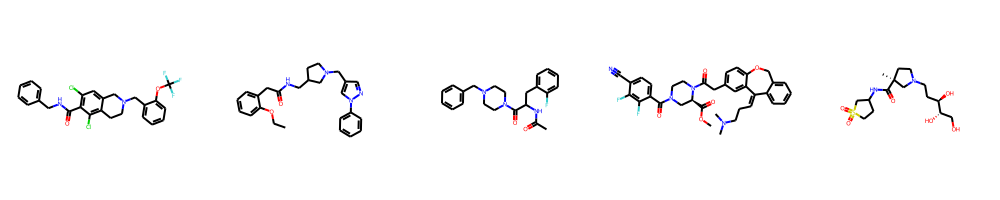
Random molecules
This endpoint returns a specified number of random molecules in SMILES format from a random selection of databases.
# Define the URL and headers
url = MY_URL + '/random_molecule'
params = {'n_mols': 5}
# Make the GET request
response = requests.get(url, headers=headers, params=params)
pprint(response.json())
[ │ 'O=C(NCc1ccccc1)c1c(Cl)cc2c(c1Cl)CCN(Cc1ccccc1OC(F)(F)F)C2', │ 'CCOc1ccccc1CC(=O)NCC1CCN(Cc2cnn(-c3ccccc3)c2)C1', │ 'CC(=O)NC(Cc1ccccc1F)C(=O)N1CCN(Cc2ccccc2)CC1', │ 'COC(=O)[C@H]1CN(C(=O)c2ccc(C#N)c(F)c2F)CCN1C(=O)Cc1ccc2c(c1)/C(=C\\CCN(C)C)c1ccccc1CO2', │ 'C[C@]1(C(=O)NC2CCS(=O)(=O)C2)CCN(CC[C@@H](O)[C@@H](O)CO)C1' ]
Available Databases
# Define the URL and headers
url = MY_URL + '/available_dbs'
# Make the GET request
response = requests.get(url, headers=headers)
pprint(response.json())
{ │ 'available_dbs': [ │ │ 'MCULE-FULL', │ │ 'ZINC15', │ │ 'SYNPLE', │ │ 'ENAMINE-CARBOXYLIC', │ │ 'EXPLORE-ENUMERATED', │ │ 'EXPLORE-DIVERSE', │ │ 'ENAMINE-REAL', │ │ 'CHEMRIYA', │ │ 'MCULE-IN-STOCK' │ ] }
Search
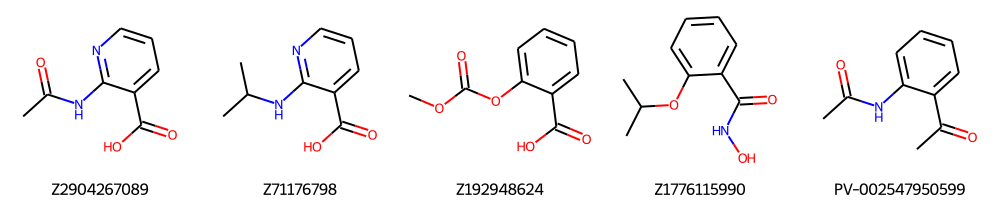
Simple Search
params = {
"search_input": "CC(=O)Oc1ccccc1C(=O)O", # SMILES of the query molecule
"search_type": 'espsim_shape', # CHEESE Search type
"n_neighbors": 5,
"search_quality": "fast",
"db_names": ["ENAMINE-REAL"],
}
response = requests.get(MY_URL + "/molsearch_simple", params=params, headers=headers)
pprint(response.json())
{ │ 'smiles': [ │ │ 'CC(=O)NC1=NC=CC=C1C(=O)O', │ │ 'CC(C)NC1=NC=CC=C1C(=O)O', │ │ 'COC(=O)OC1=CC=CC=C1C(=O)O', │ │ 'CC(C)OC1=CC=CC=C1C(=O)NO', │ │ 'CC(=O)NC1=CC=CC=C1C(C)=O' │ ], │ 'id': ['Z2904267089', 'Z71176798', 'Z192948624', 'Z1776115990', 'PV-002547950599'] }
smiles = response.json()["smiles"]
ids = response.json()["id"]
Draw.MolsToGridImage([Chem.MolFromSmiles(mol) for mol in smiles], molsPerRow=5, legends=ids)
Advanced Search
import requests
params = {
"search_input": "CC(=O)Oc1ccccc1C(=O)O",
"search_type": 'espsim_shape',
"n_neighbors": 5,
"search_quality": "fast",
"db_names": ["ZINC15"],
"descriptors": True,
"properties": True,
"filter_molecules": False,
"order_molecules": False
}
response = requests.get(MY_URL + "/molsearch", params=params, headers=headers)
pprint(response.json().keys())
dict_keys(['remarks', 'canonicalized_query', 'neighbors', 'query_properties', 'search_info'])
{ │ 'remarks': '', │ 'canonicalized_query': 'CC(=O)Oc1ccccc1C(=O)O', │ 'neighbors': [ │ │ { │ │ │ 'smiles': 'CC(=O)Oc1ccccc1C(=O)[O-]', │ │ │ 'zinc_id': 'ZINC15 : 53', │ │ │ 'embedding_distance': 0.2928953766822815, │ │ │ 'properties': {...} │ │ }, │ │ { │ │ │ 'smiles': 'CC(=O)Oc1ccccc1C(N)=O', │ │ │ 'zinc_id': 'ZINC15 : 404835', │ │ │ 'embedding_distance': 0.30397266149520874, │ │ │ 'properties': {...} │ │ }, │ │ { │ │ │ 'smiles': 'CC(=O)Oc1ccccc1C(N)=S', │ │ │ 'zinc_id': 'ZINC15 : 72284420', │ │ │ 'embedding_distance': 0.3062485158443451, │ │ │ 'properties': {...} │ │ }, │ │ { │ │ │ 'smiles': 'CC(=O)Nc1ccncc1C(=O)[O-]', │ │ │ 'zinc_id': 'ZINC15 : 3163078', │ │ │ 'embedding_distance': 0.30769655108451843, │ │ │ 'properties': {...} │ │ }, │ │ { │ │ │ 'smiles': 'CC(=O)Oc1ccccc1C(C)=O', │ │ │ 'zinc_id': 'ZINC15 : 137705', │ │ │ 'embedding_distance': 0.3079259395599365, │ │ │ 'properties': {...} │ │ } │ ], │ 'query_properties': { │ │ 'smiles': 'CC(=O)Oc1ccccc1C(=O)O', │ │ 'zinc_id': '', │ │ 'properties': { │ │ │ 'absorption': {...}, │ │ │ 'excretion': {...}, │ │ │ 'toxicity': {...}, │ │ │ 'distribution': {...}, │ │ │ 'metabolism': {...}, │ │ │ 'basics': {...} │ │ } │ }, │ 'search_info': { │ │ 'query_embedding_time': 0.37433314323425293, │ │ 'search_time': 0.45039892196655273, │ │ 'filter_time': 5.1021575927734375e-05, │ │ 'sorting_time': 1.2636184692382812e-05, │ │ 'property_prediction_time': 0.06553077697753906, │ │ 'total_time': 0.8903264999389648 │ } }
One result molecule json with properties and descriptors looks like this:
{ │ 'smiles': 'CC(=O)Oc1ccccc1C(=O)[O-]', │ 'zinc_id': 'ZINC15 : 53', │ 'embedding_distance': 0.2928953766822815, │ 'properties': { │ │ 'absorption': { │ │ │ 'caco2_wang': -4.235, │ │ │ 'lipophilicity_astrazeneca': -0.414, │ │ │ 'solubility_aqsoldb': -2.315, │ │ │ 'bioavailability_ma': 0.908, │ │ │ 'hia_hou': 0.989, │ │ │ 'pgp_broccatelli': 0.0, │ │ │ 'clogp': -0.0246 │ │ }, │ │ 'excretion': { │ │ │ 'clearance_hepatocyte_az': 86.346, │ │ │ 'clearance_microsome_az': 55.043, │ │ │ 'half_life_obach': 0.43 │ │ }, │ │ 'toxicity': {'ld50_zhu': 1.81, 'ames': 0.003, 'dili': 0.021, 'herg': 0.0}, │ │ 'distribution': {'ppbr_az': 45.823, 'vdss_lombardo': 0.155, 'bbb_martins': 0.985}, │ │ 'metabolism': {'cyp2c9_veith': 0.046, 'cyp2d6_veith': 0.0, 'cyp3a4_veith': 0.0}, │ │ 'basics': { │ │ │ 'molecular_weight': 179.03498, │ │ │ 'formal_charge': -1.0, │ │ │ 'heavy_atoms': 13.0, │ │ │ 'h_bond_acceptors': 4.0, │ │ │ 'h_bond_donor': 0.0, │ │ │ 'rotatable_bonds': 2.0, │ │ │ 'num_of_rings': 1.0, │ │ │ 'molar_refractivity': 42.0815, │ │ │ 'number_of_atoms': 13.0, │ │ │ 'topological_surface_area_mapping': 66.43 │ │ } │ } }
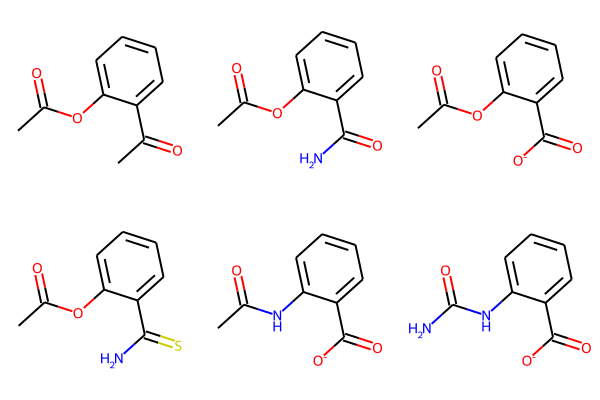
Advanced Search with filtering and ordering
import requests
params = {
"search_input": "CC(=O)Oc1ccccc1C(=O)O",
"search_type": 'espsim_shape',
"n_neighbors": 10,
"search_quality": "fast",
"db_names": ["ZINC15"],
"descriptors": True,
"properties": True,
"filter_molecules": True,
"order_molecules": True,
"filtering": ["PAINS", "Murcko scaffold hop"], # Using filtering to filter out PAINS and to get only scaffold hops
"ordering": ["Morgan Tanimoto"] # Sorting results based on Morgan Fingerprints after doing cheese search
}
def just_smiles(advanced_response):
return [r["smiles"] for r in advanced_response["neighbors"]]
response = requests.get(MY_URL + "/molsearch", params=params, headers=headers)
pprint(just_smiles(response.json())) # note: we searched 10 molecules but got 6 because of the filtering (its better to increase the number of neighbors)
[ │ 'CC(=O)Oc1ccccc1C(C)=O', │ 'CC(=O)Oc1ccccc1C(N)=O', │ 'CC(=O)Oc1ccccc1C(=O)[O-]', │ 'CC(=O)Oc1ccccc1C(N)=S', │ 'CC(=O)Nc1ccccc1C(=O)[O-]', │ 'NC(=O)Nc1ccccc1C(=O)[O-]' ]
smiles = just_smiles(response.json())
Draw.MolsToGridImage([Chem.MolFromSmiles(mol) for mol in smiles], molsPerRow=3)
Search Array
Searching a list of molecules
import requests
params = {
"search_input": ["CC1=CN(C)N=C1", "CNC1=CC=CC=C1", "CCN1C=CN=C1"],
"search_type": 'espsim_shape',
"n_neighbors": 5,
"search_quality": "fast",
"db_names": "ZINC15",
"descriptors": False,
"properties": False,
"filter_molecules": False
}
response = requests.get(MY_URL + "/molsearch_array", params=params, headers=headers)
pprint(response.json(), max_depth=3)
{ │ 'CC1=CN(C)N=C1': { │ │ 'CC1=CN(C)N=C1': {'remarks': '', 'canonicalized_query': 'Cc1cnn(C)c1', 'neighbors': [...]}, │ │ 'search_info': { │ │ │ 'query_embedding_time': 0.43999290466308594, │ │ │ 'search_time': 0.17154765129089355, │ │ │ 'filter_time': 4.3392181396484375e-05, │ │ │ 'sorting_time': 0.009363174438476562, │ │ │ 'property_prediction_time': 0, │ │ │ 'total_time': 0.6209471225738525 │ │ } │ }, │ 'CNC1=CC=CC=C1': { │ │ 'CNC1=CC=CC=C1': {'remarks': '', 'canonicalized_query': 'CNc1ccccc1', 'neighbors': [...]}, │ │ 'search_info': { │ │ │ 'query_embedding_time': 0.3788025379180908, │ │ │ 'search_time': 0.08669018745422363, │ │ │ 'filter_time': 4.506111145019531e-05, │ │ │ 'sorting_time': 0.010756254196166992, │ │ │ 'property_prediction_time': 0, │ │ │ 'total_time': 0.47629404067993164 │ │ } │ }, │ 'CCN1C=CN=C1': { │ │ 'CCN1C=CN=C1': {'remarks': '', 'canonicalized_query': 'CCn1ccnc1', 'neighbors': [...]}, │ │ 'search_info': { │ │ │ 'query_embedding_time': 0.3099024295806885, │ │ │ 'search_time': 0.06690430641174316, │ │ │ 'filter_time': 4.2438507080078125e-05, │ │ │ 'sorting_time': 0.008794546127319336, │ │ │ 'property_prediction_time': 0, │ │ │ 'total_time': 0.38564372062683105 │ │ } │ } }
Batch Search
import requests
params = {
"search_input": [
"CC(=O)NC1=NC=CC=C1C(=O)O",
"CC(C)NC1=NC=CC=C1C(=O)O",
"COC(=O)OC1=CC=CC=C1C(=O)O",
"CC(C)OC1=CC=CC=C1C(=O)NO",
"CC(=O)NC1=CC=CC=C1C(C)=O",
],
"search_type": "espsim_shape",
"n_neighbors": 10,
"search_mode": "batch", # or "centroid"
}
response = requests.get(MY_URL + "/batch_search", params=params, headers=headers)
smiles = response.json()["smiles"]
ids = response.json()["id"]
Draw.MolsToGridImage(
[Chem.MolFromSmiles(mol) for mol in smiles], molsPerRow=5, legends=ids
)
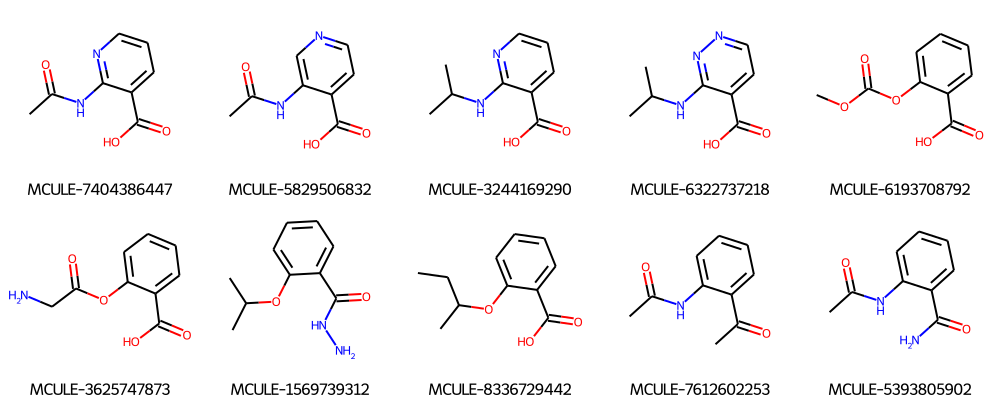
Embeddings (On-Prem users only)
You need an on-prem installation of CHEESE to use this endpoint. In public API, this endpoint will return an error.
params = {
"search_input": ["Fc1ccccc1", "Clc1ccccc1", "Brc1ccccc1"],
"save_embs": False,
}
response = requests.get(MY_URL + "/embeddings", params=params, headers=headers)
pprint(response.json().keys())
pprint(np.array(response.json()["espsim_shape"]).shape)
pprint(np.array(response.json()["espsim_shape"][0])) # embedding of the first molecule
dict_keys(['morgan', 'espsim_electrostatic', 'espsim_shape', 'active_pairs'])
(3, 256)
array([-0.271, 0.028, -0.28 , 0.089, -0.067, -0.005, -0.045, -0.024, │ │ 0.126, -0.162, -0.027, -0.163, 0.142, 0.006, -0.033, -0.069, │ -0.111, 0.045, 0.01 , 0.206, 0.006, 0.137, 0.056, 0.012, │ -0.033, -0.123, 0.013, -0.008, 0.076, 0.116, -0.274, -0.067, │ │ 0.042, -0.113, 0.017, -0.023, 0.085, -0.029, -0.059, 0.078, │ │ 0.091, -0.045, -0.034, 0.053, 0.05 , 0.119, -0.018, 0.108, │ │ 0.013, -0.015, -0.021, 0.02 , -0.086, 0.16 , 0.149, -0.174, │ -0.047, 0.241, -0.133, 0.02 , -0.265, -0.009, -0.043, -0.118, │ │ 0.112, 0.043, 0.049, -0.001, 0.042, 0.007, -0.031, 0.145, │ -0.07 , 0.048, 0.01 , -0.016, 0.039, -0.027, -0.202, 0.064, │ │ 0.044, -0.077, 0.005, -0.07 , -0.09 , 0.276, -0.047, 0.189, │ │ 0.08 , -0.094, 0.075, -0.047, 0.142, -0.242, -0.117, -0.04 , │ │ 0.177, 0.111, 0.156, -0.015, -0.032, 0.197, -0.003, -0.006, │ -0.064, -0.045, -0.051, 0.071, 0.043, 0.048, -0.103, 0.036, │ -0.078, 0.025, 0.139, -0.055, -0.014, 0.064, -0.125, 0.052, │ -0.001, 0.046, 0.02 , 0.041, 0.009, 0.025, 0.142, -0.322, │ │ 0.001, 0.046, -0.113, 0.143, 0.126, 0.045, 0.124, -0.016, │ -0.098, -0.13 , 0.23 , 0.013, -0.228, -0.069, -0.088, -0.022, │ │ 0.025, 0.011, -0.131, 0.222, -0.007, -0.092, 0.023, 0.069, │ -0.011, -0.042, 0.101, -0.056, -0.079, 0.152, -0.027, -0.012, │ │ 0.03 , 0.147, 0.006, -0.117, 0.081, -0.115, 0.101, 0.121, │ │ 0.101, -0.072, 0.011, -0.002, 0.003, -0.192, -0.024, 0.135, │ -0.027, 0.01 , 0.047, -0.116, -0.058, 0.107, 0.119, 0.002, │ -0.18 , -0.054, 0.003, -0.021, 0.031, 0.036, -0.038, -0.121, │ │ 0.145, -0.038, -0.015, -0.015, 0.028, -0.05 , -0.104, 0.146, │ -0.093, -0.056, -0.149, -0.074, 0.126, -0.07 , -0.112, 0.001, │ -0.098, -0.026, -0.007, -0.125, -0.003, 0.066, -0.081, -0.035, │ -0.09 , -0.166, -0.026, -0.114, -0.19 , -0.07 , 0.032, 0.05 , │ -0.015, -0.022, -0.128, 0.041, -0.179, 0.038, -0.161, -0.05 , │ -0.104, 0.176, -0.028, -0.117, 0.111, -0.145, 0.166, 0.226, │ -0.062, -0.019, 0.039, 0.006, 0.056, 0.138, -0.072, -0.022, │ │ 0.052, -0.122, 0.157, -0.012, -0.048, 0.122, -0.02 , -0.022])
Embeddings of lots of molecules
[ │ 'N#CCCN1N=C(c2ccc(OCc3ccccc3)cc2)OCC1=O', │ 'CC(C)C[C@H](NC(=O)c1cn(Cc2ccccc2)nn1)B(O)O', │ 'Cl.NCC(=O)CCC(=O)OCc1ccccc1', │ 'CC(C)[C@H](NC(=O)[C@H](C)N)C(=O)N1CCC[C@H]1C(=O)N(C1CCCCC1)[C@@H](C)C(=O)N[C@@H](Cc1ccc(O)cc1)C(=O)O', │ 'Nc1cccc(CP(=O)(O)CC(CCC(=O)O)C(=O)O)c1', │ 'O=C(Cc1ccc(OCc2ccccc2)cc1)N[C@@H](CCS)Cc1c[nH]c2ccccc12', │ 'COc1ccc2c(c1)C(=O)CC(CCN1CCC3(CC1)NCNC3=O)C2', │ 'COC(=O)c1cc(C(O)CN2CCN(c3ccccc3OC)CC2)ccc1OC', │ 'O=C(O)CCCOc1ccccc1-c1cc2cc(C(=O)NC(c3ccccc3)c3ccccc3)ccc2o1', │ 'N/C(=C\\C(=O)c1ccc(Cl)cc1)C(=O)O' ]
params = {
"search_input": my_smiles,
"save_embs": True,
"search_type": "all",
"dest": "/data/my_embeddings" # save into directory (faster than sending embeddings in json), creates if doesnt exist
}
response = requests.get(MY_URL + "/embeddings", params=params, headers=headers)
pprint(response.json(), max_length=5)
{'message': 'Success ! You can find computed embeddings in : /data/my_embeddings'}
Centroid Embeddings (On-Prem users only)
You need an on-prem installation of CHEESE to use this endpoint. In public API, this endpoint will return an error.
This API call retrieves embeddings of database cluster centroids.
import requests
params = {
"db_name": "ZINC15",
"search_type": 'espsim_shape',
"centroid_mols": False, # whether to output representative molecules for the clusters
"save_embs": True,
"dest": "/data/my_centroid_embeddings" # save into directory (faster than sending embeddings in json), creates if doesnt exist
}
response = requests.get(MY_URL + "/centroid_embeddings", params=params, headers=headers)
(26403, 256)
Similarity (On-Prem users only)
You need an on-prem installation of CHEESE to use this endpoint. In public API, this endpoint will return an error.
Pairwise similarity
params = {
"smiles1": "Fc1ccccc1",
"smiles2": "Clc1ccccc1",
"similarity_metric": "all",
"distance_type": "euclidean"
}
response = requests.get(MY_URL + "/similarity", params=params, headers=headers)
pprint(response.json())
{ │ 'morgan': 1.3493338152007586, │ 'espsim_electrostatic': 0.7924910655564171, │ 'espsim_shape': 0.3191955858476541, │ 'active_pairs': 0.7519632147430073 }
params = {
"smiles1": "Fc1ccccc1",
"smiles2": "Clc1ccccc1",
"similarity_metric": "all",
"distance_type": "cosine" # cosine is from 0 to 1
}
response = requests.get(MY_URL + "/similarity", params=params, headers=headers)
pprint(response.json())
{ │ 'morgan': 0.443562888451121, │ 'espsim_electrostatic': 0.21610830917440194, │ 'espsim_shape': 0.019051098635778363, │ 'active_pairs': 0.060692261872934083 }
Similarity Matrix
params = {
"smiles": [
"CC(=O)NC1=NC=CC=C1C(=O)O",
"CC(C)NC1=NC=CC=C1C(=O)O",
"COC(=O)OC1=CC=CC=C1C(=O)O",
"CC(C)OC1=CC=CC=C1C(=O)NO",
"CC(=O)NC1=CC=CC=C1C(C)=O",
],
"similarity_metric": "espsim_shape",
"distance_type": "cosine",
}
response = requests.get(MY_URL + "/similarity_matrix", params=params, headers=headers)
pprint(np.array(response.json()[params["similarity_metric"]]))
array([[1.110e-16, 4.405e-02, 7.192e-02, 1.027e-01, 3.978e-02], │ [4.405e-02, 1.110e-16, 9.728e-02, 6.456e-02, 7.657e-02], │ [7.192e-02, 9.728e-02, 0.000e+00, 9.438e-02, 9.444e-02], │ [1.027e-01, 6.456e-02, 9.438e-02, 0.000e+00, 9.449e-02], │ [3.978e-02, 7.657e-02, 9.444e-02, 9.449e-02, 0.000e+00]])
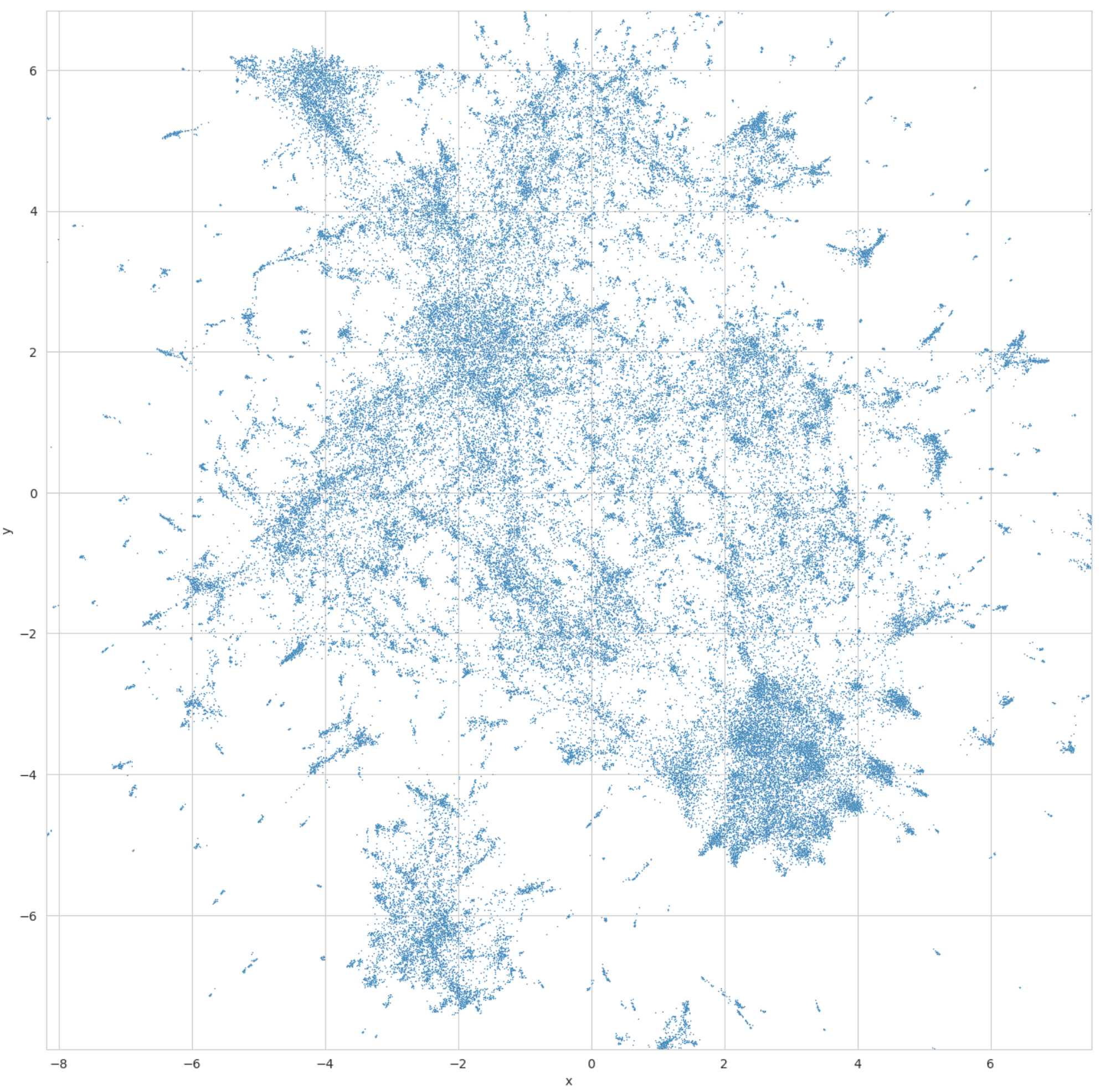
Visualisation (On-Prem users only)
You need an on-prem installation of CHEESE to use this endpoint. In public API, this endpoint will return an error.
Visualisation command works the same as embeddings command, but returns 2D coordinates intended for visualisation
params = {
"search_input": ["Fc1ccccc1", "Clc1ccccc1", "Brc1ccccc1"],
"search_type": "espsim_electrostatic",
"visualisation_method": "umap"
}
response = requests.get(MY_URL + "/visualise", params=params, headers=headers)
pprint(np.array(response.json()["espsim_electrostatic"]))
array([[8.325, 9.227], │ [8.393, 9.108], │ [8.269, 9.257]])
my_smiles = open("my_dataset.smi", "r").read().splitlines() # mix of databases
params = {
"search_input": my_smiles,
"search_type": "espsim_electrostatic",
"visualisation_method": "umap", # UMAP or PCA
"save_coordinates": True,
"dest": "/data/my_umap_coordinates"
}
response = requests.get(MY_URL + "/visualise", params=params, headers=headers)
pprint(response.json())
After loading UMAP embeddings, it looks like this.